IUPAC
When we report the chemical shift of a peak, we can use two units, according to IUPAC. The most recent recommendation I have found is:
Pure Appl.Chem., Vol.73, No.11, pp.1795–1818, 2001.
©2001 IUPAC
which defines the δ as:
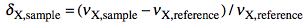
and the Ξ as:
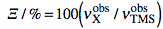
The latter term is the measured 1H frequency of TMS (diluted chloroform solution). The δ scale is limited to a single nuclide, while the Ξ scale is a unified scale valid for all nuclei. The unified scale simplifies the experimental practice for the exotic nuclei, but generates unusual figures with 6 decimal digits. I have always worked with hydrogen and carbon and have never found a chemical shift reported in Ξ units. I am perplexed because IUPAC says:
IUPAC recommends that a unified chemical shift scale for all nuclides be based on the proton resonance of TMS as the primary reference.
and also
In the future, reporting of chemical shift data as Ξ values may become more common and acceptable.
Consider that the above formula for Ξ is only apparently simple: the input values are not normally found inside NMR spectral files. Even if they are, the chemist is not used to calculate the chemical shift: he reads it. To really switch to the unified scale it would be necessary that:
- All journals require the chemical shift expressed in Ξ units.
- All spectrometers have their software updated, so the scale can be optionally be expressed in the new unit (who's going to pay?).
- The absolute frequency of 1H of TMS, measured when installing the instrument, be saved into every file.
- Some standard rule governs the previous point, so all softwares can read spectra from all instruments.
What's really good about the cited article is that it includes all the information on the subject, and we don't have to consult older recommendations (the contrary happened with the definition of JCAMP-DX for NMR, unfortunately). In the following part of this post I will discuss the old δ unit exclusively. Citing again the IUPAC article:
Unfortunately, older software supplied by manufacturers to convert from frequency units to ppm in FT NMR sometimes uses the carrier frequency in the denominator instead of the true frequency of the reference, which can lead to significant errors.
The carrier frequency is the frequency of the transmitter (the center of the spectral width). The reference frequency is the absolute frequency (in the laboratory frame) of the reference compound (TMS, just to materialize the idea). You know that TMS is usually at the far right of the spectrum, so there is a noticeable difference between the two frequencies. When the spectrum leaves the spectrometer, how can you tell which of the two values are exported with the file? I have opened a XWin-NMR file and found:
##$SFO1= 600.1324441176
##$SW= 8.26537126752216
##$SW_h= 4960.31746031746
You can verify that SFO1 = SW_h / SW. _Assuming_ that XWin-NMR follows the IUPAC convention, SFO1 is the frequency of TMS. I don't know what happens with other programs. And I don't know what happens when the user changes the scale reference. According to the rules, if he shifts the scale of even a minimal quantity, let's say 0.001 ppm, the last digits of SFO1 should change.
I know for sure that it is not so with iNMR, which is wrong. iNMR accepts the frequency value found into the original file and keeps it constant (unless the user changes it explicitly). It is possible to recalculate the reference value every time that the TMS position is redefined, but it's almost a paradox. If the users says that the scale is not correct, then other parameters may also be wrong. In order to follow the rule, the program must perform a calculation based on dubious values. If I don't follow the rule, in the common case that the scale is shifted by 0.1 ppm or less, I know to make an error in the order of 10-7. In practice an error of less than 10-4 can be tolerated. Today I prefer to introduce this minimal error, instead of altering the value for the spectrometer frequency. The user is free, however, to set both the value for the spectrometer frequency and the TMS position. Tomorrow, if I change my mind, I can make the process automatic.
posted by old swan @ 7:16 PM
0 comments
![]()

0 Comments:
Post a Comment
<< Home