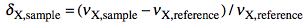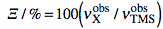NMR means nuclear magnetic resonance. It' s an analytical technique by which atomic nuclei and, indirectly, their surroundings, are revealed. It's called magnetic because a magnet is used to enhance the (otherwise negligible) energy of the nuclei. It's a resonance phenomenon because the nuclei, when excited by a wave of the right frequency, respond with an analogue wave. The NMR experiment is conceptualized in a rotating coordinate system. For the moment being you just have to pretend it is a plain Cartesian system of coordinates xyz. A radio-frequency pulse tilts the magnetization of the nuclei, initially in the z direction, in the xy plane for detection. Here each nucleus rotates at its own resonance frequency while two detectors sample the total magnetization at regular intervals along the x and y axes. The regular interval is called dwell time. The measured intensities along the x axis are called the real part of the spectrum and intensities along y form the imaginary part. Real and imaginary are just names. They could have been called right and left or red and white and it would have been the same (or better). This is the main difference indeed between an NMR instrument and a hi-fi tuner. It probably serves to justify the price difference. You have to realize that the real and imaginary parts are both true experimental values of the same importance. These intensities are stored on a hard disk in the same time order in which they are sampled (i.e. chronologically). A couple of a real and an imaginary values, collected at the same time, constitute a complex point. iNMR normally displays only the real part of the spectrum. A complex spectrum is like a vector in physics. It can be characterized by its x and y components or by its magnitude (amplitude) and direction. iNMR lets you display the magnitude of the spectrum if you want. The direction of a complex point is called phase. The so called "phase correction" is a process which mixes the real (x) and imaginary (y) components. A radio-frequency wave has frequency, amplitude and phase. Thus complex numbers are the natural choice to describe a RF signal. In an older experimental scheme a single detector measures the magnetization along both axes. In this case the sampling cannot be simultaneous. It is in fact sequential. In this case the spectrum is known as real only. Actually it can (and is) manipulated just as a normal simultaneous, complex, spectrum. A simple ad hoc correction is needed when transforming the spectrum in the "frequency domain".
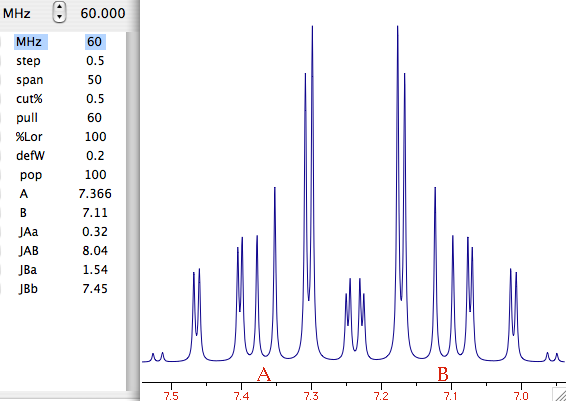
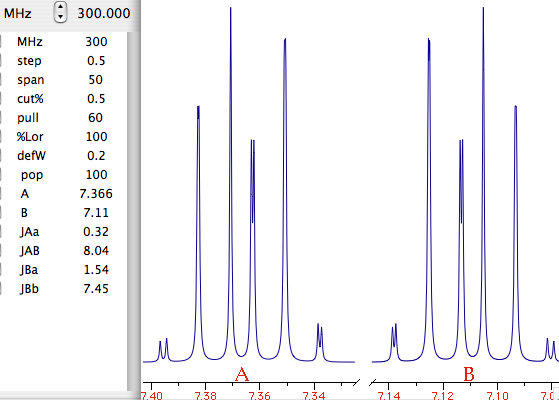
Let's explain what this last term means. What we have been speaking of up to know is a not very meaningful function of time called FID (free induction decay) (induction is another way to refer to magnetization). It is not very meaningful because all the nuclei resonates at the same time. If there were me and Pavarotti singing together it would be easy for you to discriminate our single contribution to the choir. But you are not trained to discriminate among a collection of atomic nuclei resonating together. The Fourier transformation (FT) is a mathematical tool that separates the contribution of each nucleus by its resonance frequency. The FID is a function of time, while the transformed spectrum is a function of frequency. The FT requires a computer and this is why you need a computer and a software application if you want to do some NMR. At first sight it may seem that the function of frequency should extend between - and +. Actually the sampling theorem states it only has to be calculated in the interval from 0 (included) to Ny (excluded). Ny = Nyquist frequency = sampling rate = reciprocal of the dwell time. Let's take a pause. What's the angle whose sine is 1? My pocket calculator says: 90°. I say: 90°±n360°. Who is right? Both! Coming back to our NMR experiment, suppose a signal is so fast that it rotates by exactly 360° during the dwell time. The two detectors will see it always in the same position, so they will believe it simply doesn't move (it has zero frequency). The same happens with four different signals which rotate by -350°, 10°, 370° and 730° during the dwell time. There is absolutely no way to tell which is which, unless you shorten the dwell time (a common experimental practice). A final case: you have two signals A and B and A moves of 361° respect to B each sampling interval. You will get the impression that A is moving only of 1° each time. In conclusion, the maximum difference in frequency that can be detected is = number of cycles / time interval = 1 / dwell time = Nyquist. q.e.d. In NMR this quantity is called spectral width. All the books report different expressions for the Nyquist frequency and the spectral width. One day we should open a discussion on the subject.
On a purely mathematical basis it doesn't matter how large the actual frequency range you have to record. You can pretend the range starts at zero and extend up to comprise the maximum signal separation. In practice detectors work in the low-frequency range. So you have technical limitations, and this is only the first one. The resonance frequencies today are approaching the GHz. The frame of reference also rotates at a similar frequency, so the apparent frequency is in the range of KHz. With this reduced frequency the dwell time needs to be in the order of milliseconds. The problem is that you need to filter out all other frequencies because they contain nasty noise (didn't I say it was an hi-fi matter?). So we need the rotating frame to move from the ideal world of theory and to become a practical reality. How is the rotating frame accomplished experimentally? A detector receives two signals, one coming from the sample under study and another which is a duplicate of the exciting frequency. The detector actually detects the difference between the two frequencies. To fully exploit the power of the pulse, the transmitter is put at the centre of the spectrum. Signals falling at the left of it appear as negative frequencies. (well, here it is not important if you use the delta or the tau scale and if they are positve or negative; only the concept matters). We have said that the spectrum begins at zero. In fact, if you perform a plain FT, the whole left side would appear shifted by the Nyquist frequency, then to the right of the right side!
The FT and its inverse show a number of interesting properties. The first one predicts that, if you complex-conjugate the FID, the transformed spectrum will be the mirror image of the original spectrum. In fact, if you invert the y component of a vector, you obtain its image across a mirror put along the x axis. Anything rotating counter-clockwise (positive frequency) will appear as rotating clockwise (negative frequency) and vice versa. This mirror image is mathematically called "complex conjugate". Some spectrometers already perform this operation when acquiring. This is another reason (together with sequential acquisition) why spectra coming from different instruments require different processing.
The second property predicts that changing the sign of even points of the FID is equivalent to swapping the left half of the spectrum with the right half. Because they are already swapped and need to be put back in the correct order, you understand this is an useful property. In the beginning of the SwaN-MR era this operation was simply called "swap". A day in which I was sillier than usual I changed the name in "quadrature". A possible explanation is that spectroscopists use to say "I implement quadrature detection" instead of saying "I put the transmitter in the middle of the spectrum". Like conjugation, this operation may have already been performed by the spectrometer during acquisition; in this case don't do it a second time. A third property says that the conjugated FT of a real function is symmetrical. This property is exploited by two techniques known as zero-filling and Hilbert transformation. The exploiting is so indirect and so difficult to explain, that I'll skip the demonstration. Zero-filling means doubling the length of the FID by adding a series of zeroes to its tail. During FT, the information (signal) contained in the imaginary part will fill the empty space. As a final result, you increase the resolution of the spectrum. In fact, the spectral width is already defined by the sampling rate, so adding new points results in increasing their density and the description of details. The Hilbert transformation is the inverse trading: you first zero the imaginary part, then you reconstruct it. Programs present these two operations as different commands and books describe them with different formulae, so most people don't realize that they are two sides of the same coin. There may be cases in which you are forced to zero-fill anyway. It happens that computers prefer to apply the FT only to certain amount of data, precisely to powers of 2. E.g. they like to transform a sequence of 1024 points, but never 1000. In this case 100% of computers will automatically zero-fill to 1024 points without asking your opinion. It's not the case of being fiscal here, I advise you to keep your computer happy.
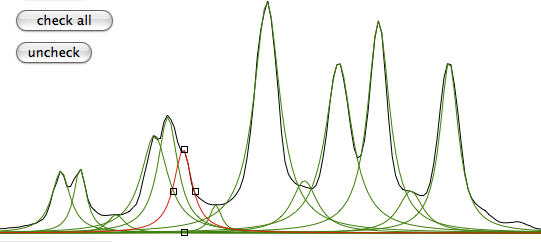
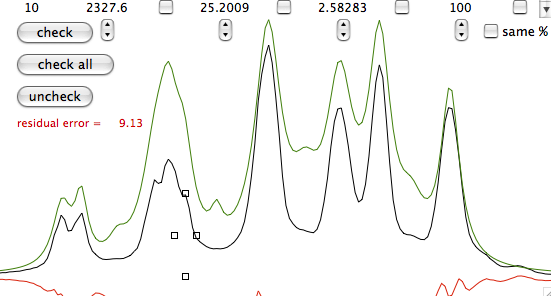
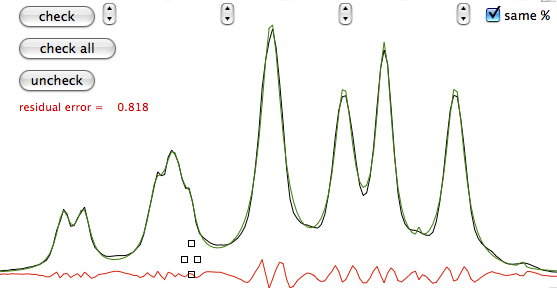
A fourth property (actually a theorem) says that multiplication in time domain corresponds to convolution in frequency domain. Convolution is crossing two functions. The product equally resembles both parents. Spectroscopists, when they dislike their spectra, use convolutions like breeders cross their cattle. Convolution certainly takes less time than crossing two animals but is still a very long operation in a spectroscopist's perception of time. So it is always preferable to perform a "multiplication in time domain" or, to save three words, "weighting". When you weight you put in practice the Heisenberg uncertainty principle. The longer the apparent life of a signal in time domain, the more resolved it will appear in frequency domain. To reach this goal you multiply the FID with a function which raises in time. At a certain point the true signal is almost completely decayed and the FID only contains noise. When you arrive there it is better to use a function which decays in time in order to reduce the noise. It's the general sort of trading between sensitivity and resolution which makes similar all spectroscopies.
A fifth property says that the total area of the spectrum is equal to the first point in the FID. This property affects both the phase and baseline characteristics of a spectrum. In fact it often happens that phase and baseline distortions are correlated. Now suppose that you have only one signal in your spectrum and that the corresponding magnetization was aligned along the y axis when the first point was sampled. The x (real) part is zero. According to the property, when you FT, half of the signal will be negative for the total area to be zero. Such a spectrum is called a "dispersion" spectrum. Normally an "absorption" spectrum is preferable, because the signal is all positive and narrower and the integral is proportional to the concentration of the chemical species. In the case described the absorption spectrum correspond to the imaginary component. In real-life cases the absorption and dispersion spectra are part in the real and part in the imaginary component. A phase correction separates them and gives the desired absorption spectrum. At last you realize why the NMR signal is recorded in two channels simultaneously! Remember it! The reverse of the coin is that the first point of the spectrum (the one at the transmitter frequency, as explained above) corresponds to the integral of the FID and, because the FID oscillates, to its average position. In theory the latter should be zero so you can decide to raise or lower the FID in order for the average to be exactly zero. In this way you reduce the artifact that the transmitter leaves at the centre of the spectrum. iNMR, by default, does not apply this kind of "DC correction". (DC stays for drift current).
A sixth property says that a time shift corresponds to a linear phase distortion in frequency domain and vice versa. Because you can' t begin acquisition during the exciting pulse (which represents time zero), you normally have to deal with this property. In fact you perform two kinds of correction. The zero-order one, described above, which affects the whole spectrum uniformly, and a first-order one, whose effect varies linearly with frequency. If in turn you want to shift all your frequencies by a non-integer number of points, the best way is to apply a first-order phase change to the FID.
Finally the Fourier transformation shares the general properties of linear functions: it commutes with multiplication by a constant and with addition. Sometimes FT alone is not enough to separate signals. In this cases the acquisition scheme is complicated with the addition of delays and pulses. During a delay, for example, two signal starting with the same phase but rotating at different frequencies lose their phase equality. A pulse has no effect on a signal aligned along its axis but rotates a signal perpendicular (in phase quadrature) to it. So you should not wonder that a suitable combination of pulses and delays can differentiate between signals. A signal is in effect a quantic transition between two energy levels A and B, caused by a RF pulse. A second pulse can move the transition to a third level C. It will be a new transition with a new frequency. This is just to show how many things can be done. In the simplest experiment the FID is a simple function of time f(t). If we introduce a delay d at any point before the acquisition the FID becomes f(d,t). Now if we run many times the experiment, each time with a different value of d, d becomes a new time variable indeed. So the expression for the FID is now f(t1, t2). t2 comes later in the experimental scheme, so it corresponds to the "t" of the simple 1-pulse experiment. What do you have on the hard disk? A rectangular, ordered set of complex points called matrix. The methods used to display it are usually borrowed by geography. If two dimensions are not enough for you, you can add a third or even a fourth one. The dimension with the highest index is said to be directly revealed because switching on the detectors is the last stage in a pulse sequence.
To go from FID(t1,t2) to Spectrum(f1, f2) you need to do everything twice. First weight, zero-fill, FT and phase correct along t2, then weight, zero-fill and phase correct along f1. Finally you can correct the baseline. The only freedom iNMR gives to you outside this scheme is that you can also correct the phase along f2 after the correction along f1. This partial lack of freedom simplifies immensely your work. Now there are good news and bad news. Bad news first. In 1-D spectroscopy you appreciated the need of acquiring the spectrum along two channels in quadrature. The books says it is required in order to put the transmitter in the middle of the spectrum. You know it is false. The true reason is that quadrature detection is needed to see the spectrum in absorption mode. The same holds for the f1 dimension. You need to duplicate everything again. Instead of using an hypercomplex point (which would need a 4D coordinate system to be conceptualised) it is customary to store points in their chronological order. You also have the real component of f1 stored in the odd rows of the matrix and the imaginary component in the even rows. The idea is that, after the spectrum is transformed and in absorption along f2, you can discard the imaginary part, because it only contains dispersion, and merge each odd row with the subsequent. After the merging your points are complex again (and the number of rows is halved). The real part is the same as before while what was the real part of even rows is now the imaginary part. After the final FT the spectrum will be phaseable along f1 (but not along f2). In case you want to perform all phase corrections at the end, do not throw away the imaginary part and process it separately. When you need to correct the phase along f2, you swap the imaginary part along f1 with the imaginary part along f2. Now the good news: iNMR does all the book-keeping for you! You just have to specify if you want to proceed with the minimal amount of data or if you prefer keeping all of it. The scheme outlined above is called Ruben-States. Another scheme exists, called TPPI. Everything was said holds, plus TPPI requires a real FT, plus it already negates even points (you have to remember of not doing it again). Just because two is not the perfect number, someone added a third scheme called States-TPPI. Fortunately it is the simplest solution, in that it requires a plain FT (no "quadrature" like Ruben-States). The most ancient (and probably the most useful) 2D experiment, the COSY experiment, is not phaseable because it is even older than the oldest scheme. So you don't need to bother with all these things. Just switch to magnitude representation. To be honest, processing a COSY experiment is certainly easier than processing a 1D proton spectrum. There is also no need to correct the baseline and less sense in measuring integrals. Another widely used protocol is echo-antiecho. It's slightly more complicated and had the merit of allowing new phase-sensitive experiments, starting with HSQC (hetero-nuclear correlation detected through the hydrogen magnetization).
If this long speech was too complicated for your tastes, it doesn't mean you cannot become an expert in NMR processing. Simply playing with iNMR, by trial and error, observing the effect of each option and pouring a small dose of common sense you can still become a wizard.